First, do no harm – making medical devices
The balance between regulation and medical device innovation
Material safety testing regulations – such as those outlined by the UK’s Medicines and Healthcare products Regulatory Agency (MHRA), the Food and Drug Administration (FDA) in the US and the European Medicines Agency – rightly mandate rigorous evaluation of medical device materials for biocompatibility, durability and compatibility with physiological environments.
Compliance with these regulations necessitates extensive testing, often involving animal studies. However, these can be time-consuming, resource-intensive, potentially unrepresentative and ethically challenging. So, when considering the cost for testing and time required for registration, only established suppliers seem equipped to handle the challenge.
These challenges become even more apparent as global regulations for material and chemical safety continue to be enhanced. Although well intentioned, these have raised difficult questions concerning supply continuity for some medical device providers. A constant balance between obligations for safety and operational margins have forced some hard decisions when it comes to the UK/EU market.
The cost of conformance
The estimated extractable testing cost range for a single-material change to a medium-high complexity class III marketed medical device can vary between €300k-€1.5mln, with this figure not insuring against the need to repeat this testing periodically to maintain conformance.
In addition, more standardised materials compliance to European and US policies ranges from €1,000-€20k, and traditional biocompatibility testing – depending on region and complexity – can vary between €2,000-€100k. This leaves small-to-medium manufacturers seeking more effective methods to meet these enhanced requirements.
Without technology-based solutions, we face the risk of supply shortages as the UK only represents approximately 10% of the global medical device market, with the US accounting for 46.6%.
Chemical challenges
Some of the most challenging commitments to chemical safety have come from EU-MDR and FDA 21CFR guidelines. These standards require devices with any form, or duration, of contact with patients to be evaluated for material, chemical and biological safety according to ISO10993-1 and ISO10993-18.
Coupled with enhanced minimum standards of analytical excellence – as governed by ISO17025, ISO14971 and ISO13485 – regulators are now requesting data at the very edge of analytical resolution, which comes at a premium cost in terms of testing. A JAMA Network Open article estimated that the non-clinical stage, including chemical testing of medical device design, represents 85% of the total cost to market.
The latest challenges forcing redesign of existing and proposed medical devices are related to the use of per- and polyfluorinated substances (PFAS), bisphenol A, polychlorinated biphenyls, and bis(2-ethylhexyl) phthalate.
The continued update of the Scientific Committee on Health, Environmental and Emerging Risks’ (SCHEER), and of carcinogenic, mutagenic, reprotoxic-endocrine disrupting substances of very high concern (CMR-ED SVHC), are also increasing complexity in the medical device supply chain. If you have heard of any of these chemicals, it is likely it was a negative announcement on its health concerns in a device.
However, what is less widely understood is that most of these chemicals have been a daily component of medical devices for decades and therefore require substantial investment and time for suppliers to retrofit. The path of least resistance is just to ban or remove these articles from the market with a declared deadline. Safer? Undoubtably yes, but for the greater benefit of society, perhaps not, when we consider that sometimes the need for the device outweighs the lifetime exposure issues.
Without a stand-in, ready-approved alternative device, we go from having an exposure profile to these chemicals that could potentially prove fatal in 30 years, to one that guarantees fatality instantly by elimination – i.e. not treating the patient in an acute setting at all.
Therein comes the concept of clinical risk benefit and risk management. What risk is ascribed to exposure in possibly acute situations versus what can be provided through design changes and supply to patients?
It’s what’s inside that counts
The standard risk assessment pathway is illustrated as part of ISO10993-18 and begins with a firm understanding of the material and chemical properties of the device in patient contact. If sufficient prior data is unavailable, additional testing must be completed in vitro.
Typical in vitro studies will assess the chemical extractables and leachables in a patient exposure. This field is a specialised form of analytical chemistry, employing chemical extraction techniques such as gas chromotography-mass spectrometry/high-resolution accurate-mass and inductively coupled plasma optical emission spectroscopy to determine chemical impurities under representative extraction settings – sometimes down to the part-per-billion trace amounts.
In vitro bench studies, although conducted relatively routinely, must always be customised to the device in question.
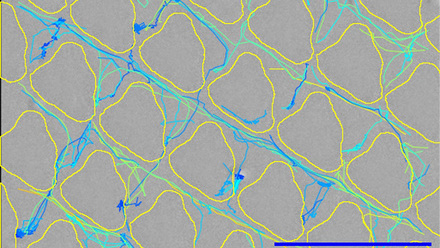
Flow studies, for example, monitor the delivery of anti-cancer treatments or radioactive microspheres via bespoke microfluidic models of tumour anatomy. They involve complex geometries or device applications, sometimes with bespoke extraction set-ups and expertise to monitor a combination of flow kinetics, patient organ architecture and device application (see image below). This can then be evaluated further with in vivo studies to support regulatory safety assessments for novel medical devices.

This level of applied in vitro evaluation was historically considered supportive of safety studies ahead of animal studies. However, happily, international regulations, in conjunction with organisations such as the 3R’s for reduction, refinement and replacement of animal testing, have started to officially recognise in vitro studies in lieu of some animal tests.
Computational modelling and simulation
In silico methods hinge on computational modelling and simulation (CMS). They offer a compelling solution to streamline material safety testing and expedite regulatory approval processes. By leveraging CMS, manufacturers can predict material behaviour, degradation kinetics and potential interactions with biological tissues with unprecedented accuracy.
Virtual testing also allows for rapid assessment of multiple material formulations, reducing the need for costly and time-consuming physical experiments, while providing robust evidence of material safety and performance.
Some approved bodies in the UK and Europe are already embracing various forms of in silico testing and evidence, plus regulators across the world are increasingly using this evidence to plug gaps, optimise clinical trial design, reduce the need for unnecessary exposure to an untested intervention/product with uncertain benefit, or reduce the need for animal testing.
The MHRA in the UK, for instance, regularly accepts CMS evidence in place of clinical studies in areas such as physiologically based pharmacokinetic modelling of absorption, distribution, metabolism and chemical substance excretion in humans. However, use of these techniques in medical devices is nascent.
One area where CMS is increasingly being developed is to assess materials in silico while capturing in situ operational conditions of the target context of use, for example, anatomical, physiological, biochemical and physiological variability.
Traditional material testing methods are often limited by their inability to capture these complex interactions between device materials and biological systems accurately. In contrast, in silico prediction models can simulate a range of physiological conditions, enabling comprehensive assessment of material biocompatibility and performance across diverse patient populations. Advanced computational techniques, such as molecular dynamics simulations and finite element analysis, provide insights into molecular-level interactions and mechanical properties.
In silico clinical trials computationally simulate medical devices in virtual environments that emulate human clinical trials. This approach benefits device designers by offering a unique and real-time platform to thoroughly iterate and optimise material and device configurations at a single patient and population level. This enables researchers to evaluate increasingly complex and realistic environmental settings when compared to traditional bench studies.
A JAMA Network Open study, titled Risk of recall associated with modifications to high-risk medical devices approved through US food and drug administration supplements, shows that modifications (e.g a change in materials) to high-risk medical devices that underwent US FDA approvals were associated with an increased risk of recall. The prevalent root cause was associated with device design (53% in Class I and 29% in All Classes)
Virtual simulations also enable rapid prototyping and evaluation of design iterations, facilitating early identification and mitigation of potential safety concerns or design flaws. By integrating material safety testing into the design process, manufacturers can minimise costly redesigns and expedite time-to-market for innovative medical devices.
Beyond regulatory compliance, in silico methods align with ethical imperatives and sustainability goals by reducing reliance on animal testing and minimising environmental impact (see image below).
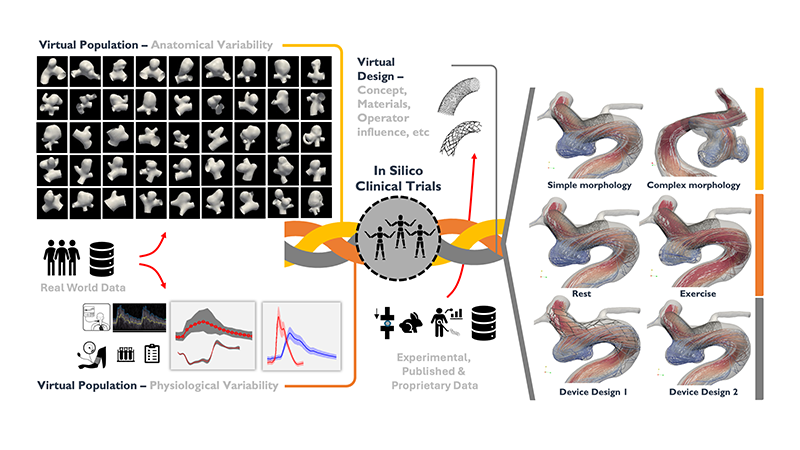
Comprehensive framework of in silico clinical trials, integrating virtual populations and designs to evaluate medical devices and treatments. Anatomical models generated from real-world data capture human diversity and physiological responses. Virtual patients undergo various medical device designs, considering factors like device concept, materials and operator influence.
The framework leverages experimental, published and proprietary data to simulate different scenarios, including morphology complexity, rest versus exercise conditions, and device comparisons, highlighting the potential of in silico trials to predict clinical outcomes and enhance medical device development
© Adapted from Sarrami-Foroushani, A. et al. In-silico trial of intracranial flow diverters replicates and expands insights from conventional clinical trials. Nat Commun 12, 3861 (2021). https://doi.org/10.1038/s41467-021-23998-w. https://creativecommons.org/licenses/by/4.0/Case study – a flow diversion neurovascular device
For example, a diverse array of materials has been developed to enhance the efficacy and safety of endovascular interventions. These materials range from flow diversion devices, particle embolics like polyvinyl alcohol particles, liquid embolics such as n-butyl cyanoacrylate, to coils and stents. They are designed to promote blood clotting within the aneurysm, reconstruct the affected vessel, or redirect blood flow away from the aneurysm sac.
Despite the significant advancements these materials represent, they of course face considerable regulatory challenges before they can be widely adopted. The FDA requires rigorous testing for safety and efficacy, often with extensive clinical trials.
Moreover, under the Humanitarian Device Exemption pathway, devices intended for conditions affecting a small patient population, highlight the complexity of navigating regulatory landscapes. These challenges underscore the balance between innovation and compliance with stringent standards, ensuring that new treatments advance care and uphold patient safety.
It is now possible, however, to computationally assess full devices for safety and performance, including their impact in achieving selective induced clotting, interaction with blood flow, vascular mechanobiology, and systemic physiology in real-world phenotypic anatomy.
Albeit it is still premature for in silico trials to replace traditional clinical trials fully, current focus is on targeting how in silico studies may enhance the type of evidence regulators currently receive and how gaps can be filled.
The potential unlocked by custom simulations of organ-device interactions, which incorporate a variety of materials and biomaterials, alongside the creation of virtual patient populations, is profound. These advancements are set to progress the evaluation process from human trials to pre-clinical assessments, thereby accelerating the pace of innovation and regulatory processes.
Future outlook
The revelations from investigations like the Implant Files and the Independent medicines & medical devices safety review underscore the reality that our understanding of safety can always be enhanced. However, this must be balanced against the necessity of providing patients with access to potentially lifesaving technologies, while ensuring their safety and maintaining supply integrity.
The integration of sophisticated analytical techniques and in vitro testing, along with advancements in CMS and in silico trials, herald unprecedented frontiers in medical device innovation and regulation. This evolution offers innovative pathways for assessing patient safety profiles early at the R&D phase by manufacturers.
From a financial and supply standpoint, in silico prototyping and safety evaluations promise significant reductions in time and costs, potentially influencing the decision to continue supplying a medical device to the market.
Nonetheless, the challenge extends beyond technology to include regulatory culture. Computational models must establish their credibility before they can be used as reliable regulatory evidence. Emerging standards from organisations like the American Society of Mechanical Engineers and the International Organization for Standardization (ISO) are shaping FDA guidelines on model-informed regulatory evidence reporting and credibility assessment.
The ongoing alignment among thought leadership organisations and practice communities, including the Avicenna Alliance, InSilicoUK and the Medical Device Innovation Consortium, is critical. This collaboration aims to foster policy harmonisation and governance through entities such as the International Medical Device Regulators Forum, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use and ISO. New companies offering advanced regulatory tools, such as Virtonomy and adsilico, support the delivery of in silico studies and regulatory evidence.
Finally, public advocacy groups and patient voices play a pivotal role, serving as a constant reminder of the imperative for change – a change that must adhere to the Hippocratic principle of Primum non nocere (First, do no harm), ensuring that economic and efficiency arguments do not override it.
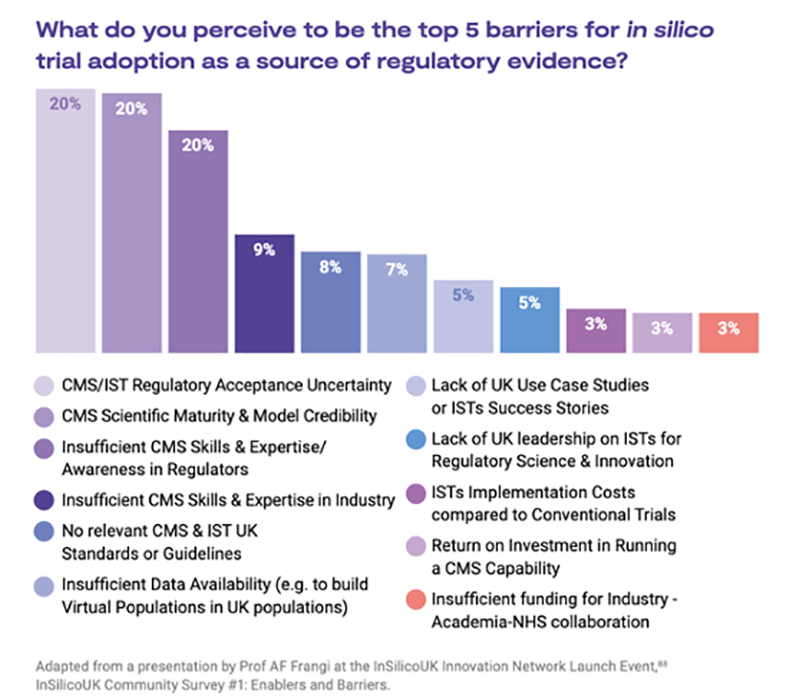
Barriers to adoption of in silico methods
Leading international efforts, such as the Avicenna Alliance, InSilicoUK and the Medical Devices Innovation Consortium, are at the forefront of advocating for the adoption of in silico computer modelling and simulation methods as a credible source of regulatory evidence.
They have identified and documented the obstacles and facilitators for implementing in silico methods. The illustration below outlines the most significant barriers as identified by a survey of over 200 regulatory scientists and practitioners.